
ГОУ ВПО томский государственный университет |
кафедра органической химии |
Карбоновые кислоты |
Строение и классификация
Соединения, содержащие группировку –СООН (карбоксильная группа), называют карбоновыми кислотами.
Карбоновые кислоты подразделяют:
- по числу карбоксильных групп- на монокарбоновые, дикарбоновые, трикарбоновые и т.д.;
- по строению радикала- на алифатические карбоновые и ароматические (бензолкарбоновые) кислоты;
Строение и свойства карбоксильной группы
Карбоксильная группа поляризована электроноакцепторными свойствами атомов кислорода и способна к ионизации в растворах:
Карбоновые кислоты, начиная с метановой- жидкости. Карбоновые кислоты в жидком состоянии ассоциированы за счет водородных связей, поэтому их температуры кипения выше, чем у спиртов с таким же числом атомов углерода:

Низшие карбоновые кислоты отличаются резким неприятным и раздражающим запахом, смешиваются с водой во всех соотношениях. Однако уже масляная кислота с водой смешивается неполностью, образуя на ее поверхности масло, откуда и получила свое название.
Производными карбоновых кислот называются соединения, которые в результате гидролиза в воде приводят к карбоновым кислотам. К производным карбоновых кислот относят: галогенангидриды, ангидриды, сложные эфиры, амиды и нитрилы, а также эфиры ортокислот:

Реакционная способность соединений к гидролизу падает в указанном направлении.
Номенклатура IUPAC
При наименованиях карбоновых кислот выделяют самую длинную цепь углерода, включающую карбоксил. Атому углерода карбоксильной группы присваивается номер 1 и от него начинается нумерация цепи. Название формируется перечислением номеров и наименований заместителей и названия углеводорода, соответствующего общему числу атомов углерода в цепи с добавлением окончания – овая кислота.
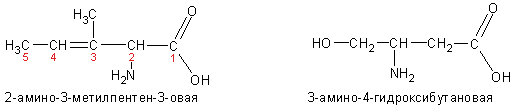
По заместительной номенклатуре кислоты могут называться как производные кислот с меньшим числом атомов углерода. Например, триметилуксусная или триметилэтановая (2,2-диметилпропановая) кислота.
Широко распространены также и тривиальные, исторически сложившиеся наименования:

Гомологический ряд дикарбоновых (диовых) кислот:

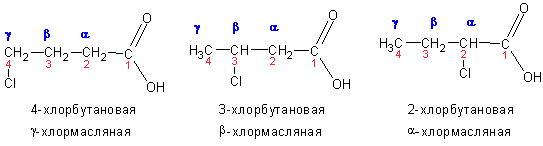
Нередко встречается способ наименования с использованием обозначения положения заместителей буквами греческого алфавита. Обратите внимание на то, что обозначение начинается с атома углерода, соседнего с карбоксильной группой:
Номенклатура длинноцепочечных карбоновых кислот, распространенных в растениях и живых организмах. Длинноцепочечные карбоновые кислоты с числом атомов углерода 16-20 очень широко распространены в живой природе. Их номенклатура имеет свои особенности. Насыщенные кислоты кроме наименований по номенклатуре IUPAC могут обозначать и тривиальными названиями. У ненасыщенных кислот также имеются тривиальные наименования. Кроме того, оба типа кислот имеют сокращенные наименования, указывающие на количество атомов углерода в молекуле и количество ненасыщенных связей. Эти данные объединены в таблице:
Число атомов углерода |
насыщенные |
ненасыщенные |
16 |
С16:0, пальмитиновая (гексадекановая) |
С16:1 пальмитолеиновая (9-гексадеценовая) |
18 |
С18:0 стеариновая (октадекановая) |
С18:1 олеиновая (9-октадеценовая С18:2 линолевая (9,12-октадекадиеновая) С18:3 линоленовая (9,12,15-октадекатриеновая) |
20 |
С20:0 арахиновая (эйкозановая) |
С20:4 арахидоновая (5,8,11,14-эйкозантетраеновая) |
Конфигурации кратных связей всех природных ненасыщенных кислот находятся в конформации цис-.
Химические свойства карбоновых кислот
В отличие от спиртов и фенолов, карбоновые кислоты обладают заметной кислотностью, хотя и значительно уступают по силе минеральным кислотам, таким, как фосфорная или соляная. Поскольку кислотность напрямую связана с легкостью отщепления протона, сила кислоты увеличивается, если отрицательный заряд аниона лучше компенсируется отрицательными индуктивными или мезомерными эффектами:

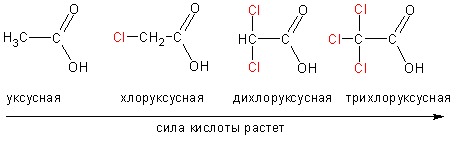
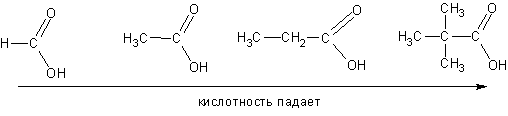
И наоборот, кислотность падает, если анион дестабилизирован электронодонорными эффектами алкильных групп:
Дикарбоновые кислоты сильнее монокарбоновых (влияние соседней карбоксильной группы с ее –I эффектом), но в ряду гомологов кислотность падает с удалением карбоксильных групп по цепи:

Тем не менее, сила большинства карбоновых кислот достаточна, чтобы вытеснить угольную кислоту из ее солей:
R-COOH + NaHCO3 → R-COONa + CO2 + H2O
Карбоновые кислоты не реагируют с солями более сильных кислот- фосфорной, соляной серной и т.д.
Значения рКа карбоновых кислот, соответствующие процессу:
R-COOH + H2O → R-COO- + H3O
находятся в пределах 4,7-4,9. Исключения составляют муравьиная кислота (рКа=3,75), а также оксо-, окси-, амино- и галогензамещенные кислоты, у которых рКа заметно ниже (сила кислоты, соответственно, больше).
Кальциевые соли практически всех карбоновых кислот в воде нерастворимы, хорошо растворимы натриевые, аммониевые и калиевые соли.
- реакции карбоксильной группы;
- реакции углеродного скелета.
1. Образование амидов и нитрилов

Амиды практически лишены основных свойств азота и водном растворе нейтральны. Сопряженная кислота ацетамида, например:

Имеет рКа~ -1,5 (это означает, что протон на ней практически не удерживается, то есть ацетамид лишен оснОвных свойств). Единственный жидкий при нормальной температуре- формамид, остальные амиды- кристаллические вещества.
Кроме дегидратации амидов, нитрилы можно получать также обменными реакциями алкилгалогенидов и цианидов калия или натрия:

Номенклатура амидов
Амиды называют по названию алкана, соответствующего числу атомов углерода в самой длинной цепи, содержащей амидогруппу, с перечислением заместителей и добавлением окончания – амид, например:

Номенклатура нитрилов
Нитрилы называют по имени алкана, соответствующего числу атомов углерода в цепи (включая углерод цианогруппы) с добавлением окончания – нитрил, например:

2. Получение ангидридов кислот
Ангидриды кислот можно рассматривать как продукты дегидратации двух молекул кислоты. При отнятии воды от карбоновых кислот (которое удается осуществить только при помощи фосфорного ангидрида Р2О5) можно получать смешанные и моноангидриды:

Другой способ получения ангидридов- реакция солей карбоновых кислот с галогенангидридами кислот:

Уксусный ангидрид- жидкость, кипящая при 140 оС, обладающая неприятным резким запахом. Ангидрида муравьиной кислоты не существует.
3. Получение галогенангидридов кислот
Хлорангидриды кислот получают классическими реакциями замены гидроксильной группы на галоген (так же, как и в случае со спиртами):

Или при реакции соли карбоновой кислоты с хлористым сульфурилом:

На рисунке показано получение хлорангидрида уксусной кислоты. Фторангидриды кислот получают из хлорангидридов при реакции с KHF2.
Номенклатура галогенангидридов
Галогенангидриды называют по названию алкана, соответствующего числу атомов углерода в самой длинной цепи, содержащей галоид, с перечислением заместителей и добавлением окончания – оил и наименования галогенида, например:

4. Получение сложных эфиров
Этерификация кислот спиртами приводит к сложным эфирам с переменным успехом.
Реакция протекает в кислой среде и обратима. Роль кислоты заключается в активации (усилении реакционной способности) карбоновой кислоты (но не спирта!) путем протонирования:

Протонированная по атому кислорода карбонильной группы кислота атакуется атомом кислорода спирта (на нем имеются две неподеленные пары электронов):

Реакция завершается переносом протона, отщеплением воды и, наконец, отщеплением самого протона (регенерация катализатора):

Поскольку все стадии этерификации обратимы, для увеличения выхода приходится отгонять из реакционной смеси продукт (эфир) или связывать воду. Обратите внимание, что в составе сложного эфира присутствует атом кислорода от спирта, а не от кислоты. Успех реакции в значительной степени зависит от строения спирта и кислоты. Так, наиболее успешно реакция протекает с короткими молекулами спиртов и кислот. Важное значение имеет то, чтобы ни спирт, ни кислота не были стерически затруднены у реакционных центров (гидроксильной и карбоксильной групп). Скорость реакции этерификации кислот спиртами растет с понижением значения рН до определенной величины, а с дальнейшим снижением рН опять падает. Связано это со все возрастающей степенью протонирования (по атому кислорода) молекулы спирта и, соответственно, потерей ей реакционной способности (атаковать карбонильный углерод).
Другим удобным способом получения сложных эфиров является реакция солей карбоновых кислот с галоидными алкилами:

При наличии ангидридов и галогенангидридов кислот наиболее удобно получать эфиры из них, так как эти реакции идут практически полностью (и необратимо). С галогенангидридами спирты реагируют по схеме:

С ангидридами реакции идут немного менее активно, но гораздо лучше, чем с самими кислотами. В реакцию берут спирт и ангидрид кислоты:

Номенклатура сложных эфиров
Сложные эфиры называют по названию радикала спирта, от которого образован сложный эфир, добавлением названия алкана, соответствующего числу атомов углерода в самой длинной цепи, содержащей карбонильную группу, с перечислением заместителей и добавлением окончания – оат , например:

Сложные эфиры низкомолекулярных карбоновых кислот и спиртов – жидкости, с приятными фруктовыми запахами (банан, вишня, груша, яблоки и т.д.). Сложные эфиры длинноцепочечных карбоновых кислот и таких же спиртов- твердые вещества, смесь которых составляет растительные и животные воски, а также и пчелиный воск. Воски защищают фрукты от неблагоприятных атмосферных воздействий, а шерсть и перья животных и птиц- от влаги и грязи. Миристилпальмитат, локализованный в углублениях костей черепа кашалота, является проводником звуков при эхолокации.
Натриевые соли длинноцепочечных карбоновых кислот используются в качестве основы для производства мыла, поскольку обладая двойственной природой, их молекулы способны эмульгировать жир и грязь, унося их с водой. Калиевые соли жирных кислот являются компонентом жидкого мыла.
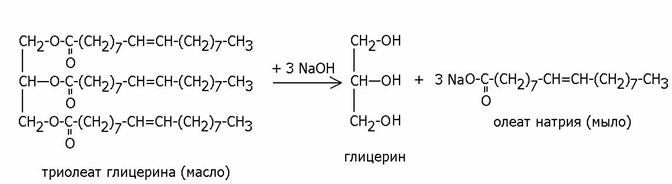
Эфиры карбоновых кислот с глицерином представляют собой жиры и масла. Если в состав сложного эфира с глицерином входят остатки насыщенных карбоновых кислот (пальмитиновой С16:0, стеариновой С18:0, арахиновой С20:0), то эфиры представляют собой твердые при нормальной температуре жиры (животные жиры и сало). Если в состав эфиров входит 2 и более остатка непредельных карбоновых кислот (пальмитолеиновой С16:1, олеиновой С18:1, линолевой С18:2, линоленовой С18:3 или арахидоновой С20:4), то такие эфиры являются жидкими при нормальной температуре маслами (например, растительные масла).

Жиры и масла с водой не смешиваются и не эмульгируют, но при обработке щелочами при нагревании омыляются, гидролизуясь до глицерина и солей карбоновых кислот, называемых мылами:
5. Получение эфиров ортокислот
Карбоновые ортокислоты в свободном виде (в отличие от фосфорной кислоты), не существуют:

Однако их орто-эфиры вполне устойчивы и могут быть получены, например, из тригалоидных алкилов и алкоголятов:

Из хлороформа, таким образом, получаются ортомуравьиные эфиры.
Условие- галоидные алкилы не должны иметь на соседнем атоме углерода ни одного атома водорода, например:

Если у a- углерода найдется атом водорода, то произойдет отщепление HCl:

Ортоэфиры можно также получить действием на нитрилы хлористым водородом в присутствии избытка спирта:

Эфиры гидролизуются по стадиям, до образования вначале сложного эфира, а уже затем он гидролизуется до кислоты и спирта.
6. Получение a-галогензамещенных кислот
При реакциях карбоновых кислот с галогеном (бром) в присутствии красного фосфора получаются a-галогензамещенные кислоты:

7. Другие реакции карбоновых кислот и их производных
Пиролиз солей карбоновых кислот приводит к алканам с тем же числом атомов углерода, что и в радикале соли (см. получение алканов).
Электролиз растворов солей карбоновых кислот приводит к образованию алканов с удвоенным числом атомов углерода по сравнению с радикалом соли.
Пиролиз кальциевых солей карбоновых кислот приводит к образованию кетонов с частичным декарбоксилированием:

Кислоты окисляются двуокисью селена SeO2 в a-кетокислоты:

Аналогичные реакции характерны и для карбонильных соединений- альдегидов и кетонов. Окисление протекает всегда по соседнему с карбонильной группой углероду.
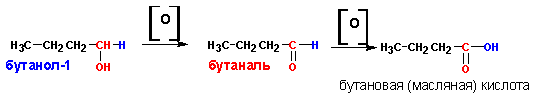
1. Окисление первичных спиртов последовательно приводит к карбоновым кислотам:
2. Окисление кетонов
Кетоны получаются в результате окисления вторичных спиртов и после жесткого окисления дают смеси кислот:

3. Окисление непредельных углеводородов
Реакции озонирования

Синтетического применения не имеет, используется только для установления места положения тройной связи. Кислоты получаются в качестве «побочных» продуктов. Однако, в некоторых случаях может являться единственным способом «мягкого» введения карбоксильной группы.
Окисление алкенов и алкинов горячим раствором КMnO4 дает аналогичный результат (см. свойства непредельных соединений).
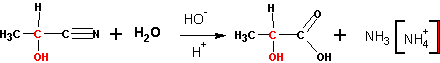
4. Омыление оксинитрилов (циангидринов)
Приводит к получению a-оксикислот:
5. Омыление нитрилов
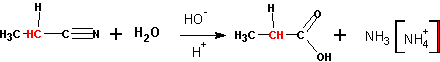
Сами нитрилы можно получать обменными реакциями цианистого калия или натрия с галоидными алкилами (см. выше также другие способы):
6. Реакции с магнийорганическими реагентами (реактивами Гриньяра)
Магнийорганические соединения перемешивают с тонкоизмельченным «сухим льдом» (твердой двуокисью углерода СО2), после чего смесь выливается в разбавленный раствор уксусной или соляной кислоты:

Если карбоксильная группа стоит при бензольном кольце, такие кислоты называют бензолкарбоновыми. Тривиальное название простейшей такой кислоты- бензойная. Бензойная кислота немного сильнее уксусной по причине отрицательного индуктивного эффекта атома углерода кольца, находящегося в состоянии sp2- гибридизации.
Сила бензолкарбоновых кислот сильно варьирует в зависимости от наличия и положения в кольце заместителей. Расположенные в орто- или пара- положении к карбоксильной группе заместители влияют на состояние атома углерода, с которым связана карбоксильная группа, и уже через него- на степень стабилизации или дестабилизации образующегося в результате отщепления протона аниона кислоты, увеличивая или уменьшая кислотность.
Заместители I рода (электронодонорные) уменьшают кислотность:

а заместители II рода кислотность увеличивают:

Напомним, что стягивание электронной плотности с карбоксильной группы облегчает отщепление протона, следовательно, увеличивает силу кислоты. Напротив, подача электронной плотности в карбоксильную группу дестабилизирует образующийся анион и он образуется труднее (протон не отсоединяется).
Большое значение имеет положение заместителя. Находясь в орто-положении к карбоксильной группе, заместители 1 рода влияют также на кислотность отрицательными индуктивными эффектами (группировки –ОН, -NH2, галогены –Х). Влияние индуктивными эффектами из пара- положения затруднено потому, что индуктивный эффект быстро затухает с увеличением расстояния.
Увеличение числа заместителей усиливает «эффект». Например, кислотность соединений меняется в показанном направлении:

В таблице приведены значения рКа для ряда замещенных бензойных кислот
рКа бензойных кислот |
|||
заместитель |
орто- |
мета- |
пара- |
3,5-динитро |
2,83 |
||
Нитро- |
2,17 |
3,45 |
3,43 |
Cl |
2,94 |
3,83 |
3,99 |
Br |
2,85 |
3,81 |
4,00 |
Me |
4,09 |
4,09 |
4,47 |
HO |
2,98 |
4,08 |
4,58 |
Из данных таблицы можно сделать вывод о том, что влияние нитрогруппы на кислотность индуктивным эффектом настолько велико, что пара- и мета- изомеры практически равны по кислотности. (При нахождении группы в мета- положении относительно карбоксильной группы мезомерный эффект на нее непосредственно не действует). Об этом же свидетельствуют и данные о кислотности орто- и пара- изомеров оксибензойной кислоты. (при нахождении группы –ОН по соседству с карбоксильной группой, последняя испытывает значительный отрицательный индуктивный эффект кислорода, отчего сила кислоты возрастает). Исходя из всего становится ясно, что предсказание силы ароматических кислот непростое занятие.
Получение бензолкарбоновых кислот
Алкилбензолы окисляются при кипячении в растворе KMnO4 до соответствующих карбоновых кислот. При окислении гомологов бензола получаются карбоновые кислоты независимо от длины и строения радикала.

Такие же результаты получаются при окислении хромовой смесью и разбавленной
азотной кислоты.
Бензолдикарбоновые кислоты (в частности, терефталевая- п-бензолдикарбоновая) используются при синтезе полимеров, при конденсации с этиленгликолем (образование сложных эфиров).

Получающиеся полимеры, называемые полиэтиленгликольтерефталатами (ПЭТ, лавсан), отличаются повышенной прочностью и светостойкостью. Из него изготавливают ткани, основу для магнитных и видеолент, а также пластиковые бутылки для пива и газированной воды.
Химические свойства дикарбоновых кислот
Как уже было сказано, сила дикарбоновых кислот (по первой ступени ионизации) выше, чем у монокарбоновых из-за влияния второй карбоксильной группы и эта сила понижается по мере удаления второй группы по цепи атомов углерода.
Однако наличие второй группы приводит к появлению еще одного эффекта. При нагревании дикарбоновые кислоты декарбоксилируют, при этом легкость удаления СО2 опять зависит от близости второй карбоксильной группы. Так, щавелевая кислота декарбоксилирует уже при 150 оС, примерно в таких же условиях теряет СО2 и малоновая кислота:
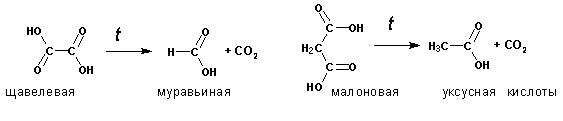
Остальные кислоты декарбоксилируют с трудом, зато при увеличении числа атомов углерода между карбоксильными группами возрастает вероятность образования циклических ангидридов в результате внутримолекулярной дегидратации. При длительном нагревании или в присутствии водоотнимающих агентов, янтарная и глутаровая кислоты образуют циклические ангидриды:
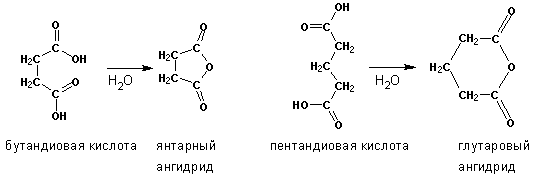
Наличие метиленовой группы малоновой кислоты между двумя карбоксильными сильно повышает кислотность ее атомов водорода. С этим связана группа реакций конденсаций производных дикарбоновых кислот (обычно- сложных эфиров), используемых в синтезе карбоновых кислот и кетонов.
В общих чертах синтезы дикарбоновых кислот не отличаются от методов синтеза монокарбоновых кислот.
Так, наиболее часто используется омыление нитрилов. Велер еще в 1824 году синтезировал щавелевую кислоту гидролизом ее динитрила- дициана:
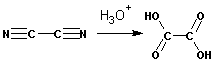
Малоновую кислоту получают из ее мононитрила- циануксусной кислоты:

Высшие дикарбоновые кислоты чаще получают окислением циклических кетонов азотной кислотой:

© khassanov, MMII-MMXVI |