Это приводит к тяжелому отставанию в умственном развитии у детей, где прежде всего патология и проявляется спустя некоторое время или сразу после рождения.
|
Молекулярные патологии |
Понятие введено Полингом в 1949 году. (Синонимы: протеинопатии)
А) ферментопатии (дефекты ферментов- фенилкетонурия и галактоземия)
Б) неферментные протеинопатии (серповидноклеточная анемия)
А) ферментные протеинопатии
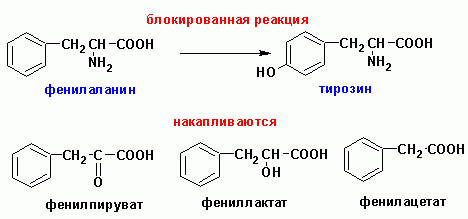
К ферментопатиям аминокислотного обмена относится
фенилкетонурия (дефект фермента фенилаланингидроксилазы),
превращающей фенилаланин в п-гидроксифенилаланин (тирозин). В результате происходит
накопление фенилпировиноградной кислоты (2-оксо-3-фенилпропановой), а также
2-гидрокси-3-фенилпропановой и фенилуксусной.
Это приводит к тяжелому отставанию в умственном развитии у детей, где прежде
всего патология и проявляется спустя некоторое время или сразу после рождения.
Галактоземия (ферментопатия углеводного обмена).
Дефект фермента галактозо-1-фосфат трансферазы. Галактозо-1-фосфат токсичен для организма. Поступающая с молоком галактоза превращается в галактозо-1-фосфат, но далее не превращается в УДФ-галактозу из-за отсутствия или неактивности выше обозначенного фермента. У грудных наблюдается замедление умственного и физического развития. Если не приостановить кормление грудью, то возможен летальный исход.
Б) Неферментные протеинопатии
Серповидноклеточная анемия.
Мутация в 6-м кодоне ДНК (замена последовательности Ц-Т-Т на Ц-А-Т) приводит к замене при трансляции (синтез бетта-цепи гемоглобина) остатка ГЛУ (глутаминовая кислота, положение 6) на остаток аминокислоты ВАЛ (валин). Замена гидрофильного остатка на гидрофобный приводит к слипанию молекул гемоглобина в ассоциаты (особенно в дезокси-форме), отчего искажается форма клетки, содержащей гемоглобин- эритроцита. Клетки при рассматривании под микроскопом имеют вид полумесяца, откуда патология получила название. Клинические симптомы впервые наблюдались в Чикаго в 1904 г., однако причина заболевания была установлена много позже. Мутировавший гемоглобин получил название HbS, или Гемоглобин Чикаго 6b GlyàVal.
Молекулярные патологии вызваны изменениями генетического кода- так называемыми МУТАЦИЯМИ. Различают ХРОМОСОМНЫЕ мутации (изменение числа хромосом) и молекулярные (ГЕННЫЕ) мутации. Последние появляются в результате одного из нескольких возможных процессов:
1. ТРАНЗИЦИЯ- или замена пар оснований
2. ДЕЛЕЦИЯ- или выпадение одной или нескольких пар
3. ВСТАВКА
4. Изменение местоположения.
МУТАГЕНЫ- факторы, вызывающие мутации, различают
А) природные
Б) чужеродные (химические вещества, в том числе лекартсва, и физические или биологические факторы- излучения вирусы и т.п.). Мутагены в окружающей среде многочисленны. Ежегодно рождается около 15 тысяч детей с дефектами генов, обусловленных лишь одними только испытаниями ядерного оружия.
Принципы лечения и профилактики молекулярных патологий
1. Исключение из пищи субстрата блокированной реакции (например, обедненная фенилаланином диета).
2. Обогащение пищи веществами, отсутствующими в результате нарушения обмена (например, при ОРОТАТАЦИДУРИИ- нарушении синтеза оротовой кислоты, являющейся предшественником пиримидиновых оснований, развивается металлобластическая анемия. Обогащение диеты цитидиловой кислотой позволяет обойти заблокированную стадию превращений).
3. Обогащение организма кофакторами при дефекте апофермента сложного фермента (назначение значительных доз витаминов и коферментов).
4. Связывание и выведение из организма токсических продуктов.
ВЕРНУТЬСЯ НА НАЧАЛЬНУЮ СТРАНИЦУ
Передача наследственной информации
|
|